




 ��ǰ������棺
��ǰ������棺��������S��ˎ���о��IJ��������Լ��s�|�о�Ҫ����ߣ��s�|�ķ������g�Լ��о��������l������Ҫ�ĸ�׃���ڌ��s�|�������������r���������s�|�о��^���Ƿ��������Ļ��A�������x����m�ķ������gҲ���P��Ҫ��
һ���s�|�ā�Դ����
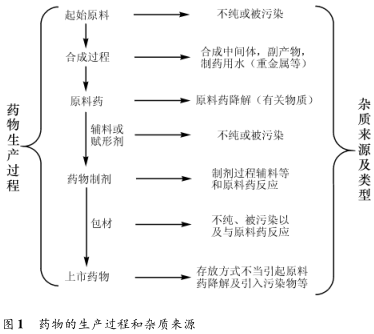
ˎ���е��s�|���܁�Դ��ˎ�����a�Լ��N�۵ȸ����h��(�D 1)������ ICHָ��ԭ�t�Ɍ�ˎ���s�|�֞��ЙC�s�|���o�C�s�|�������܄��Լ������s�|��������Ҫᘌ��ЙC�s�|�M��̽ӑ��
��ˎ���s�|�о��r���롰�|��Դ���OӋ( Quality byDesign��QbD)�����������ˎ�����a֮ǰ�������w��ˇ�ĺϳəC�ơ���ʼ���ϼ������g�w�Ļ����Y���������������aƷ���s�|�V��
�s�|��Դ�������ƶ�ˎ���s�|���Ʋ��ԵĻ��A���������ڌ������s�|��Դ�����r�����������кϳɺ����a��ˇ�е�ԇ�������g�w�����a��Ɯy���ܮa���ĝ����s�|�Լ��������H���ڵ��s�|��
��ԭ��ˎ�ϳɽY����ˎ��Ļ��Ի������mȻ���^���Է����Ѳ����С���ʾ�Y����(alerting structure)�����������a�^����ʹ�õ����о�ʾ�Y���Ļ�����t߀�迼�]���z�����ԡ�
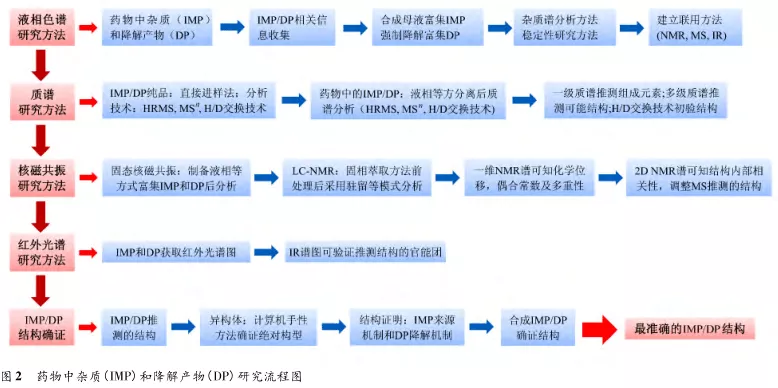
�����s�|�������
��ˎ���аl�^���У�ˎ���s�|�ķ������P�I����ˣ����s�|�о����������s�|�Y����˼·����D2���Լ����m���s�|�������g�ɘO��ؿs���s�|�о��r�g���Ƅ���ˎ���о��Ŀ��ٰlչ��
1���s�|ǰ̎�����g
�s�|��ǰ̎���ǰ��S��ˎ����Գɷ�ǰ̎�������ڵģ�Ȼ��ˎ�����s�|�ĺ���������Y���c���ɷֲ�^����˳�Ҏˎ����Գɷֵ�ǰ̎���͙z�y����(���ʼ�������ܽ��ֱ���M�� HPLC-UV ����)����һ���m����ˎ���s�|����ᘌ���ͬ�Ę�Ʒ�x��ͬ��ǰ̎�����g��
��1���z�y�`���ȵ͵Ę�Ʒ
���z�y�`���ȵ͵Ę�Ʒͨ��ʹ����������ǰ̎����ʽ������������ɫ�F�a������푑������������x�ӻ����F�����x�ӻ�Ч�ʵȡ�
�mȻ��Ҏ��������ʽ�܉�M���ճ��z�y�����������ˌ��F���͝�ȵĻ����s�|�M�п��ٺY�x�Ͷ������Ɍ����y��������ԇ���M�и�׃�������䌣���Ժ��`���ȣ�Ҳ��ʹ�Ú�-���������폛�aҺ-���������IJ��㡣
��2���͝�ȵ��s�|
�͝���s�|ǰ̎���������x��������s�|������Q�����罵��a�����Ï��ƽ���ȷ�����߽�����ĝ�ȵȣ����dz�Ҏ�Ľ��ⷽ�����������������s�|��������ɔ_�����s�|���s�|�V�о������˵õ���һ���s�|�о��C�ƣ�Ueya-ma �������һ�N���͵Ĺ��wˎ����������ƽ�_��ԓƽ�_�ų��˳�Ҋ������ʽ(���� H2O2 ����)�����ˮ�⡢�܄�����Ч���ȣ���������������C�Ƶ��خ����о���
��3������Ⱦ�x���Ę�Ʒ
��ͬ�x���в�ͬ��ʹ�×l������ˌ����s��Ʒ�M��ǰ̎�������܉����L�x����ʹ�É����������|�V�z�y������ʹ�ú��зǓ]�l���}�������࣬����ڽ���Һ�|�×l���r�����ö��SҺ��ɫ�V���g�ڵ�һ�S�������M�з��x������Ʒ��������Ʒ�h�У��ڶ��SҺ��ʹ���|�V�ɽ��ܵ��������Լ�Ó�}����ϴÓ��Ʒ�h�еĘ�Ʒ�Ķ����F�˱������Ó�}�������o�|�V��
2���s�|���x���g
�s�|��ˎ���к����^�ͣ�����ֱ�Ӝy�����o�����F�s�|�Ķ��Զ�����������ˣ������s�|�M�з��x�ԫ@ȡ�s�|�Ć�һ�ɷ֣��Ķ����F���s�|�ęz�y���������Һ��ɫ�V���g�Լ����R�����w���g�lչ�^��Ѹ�١�
��1��Һ��ɫ�V���g
��ЧҺ��ɫ�V���g(HPLC) HPLC ��������y�ķ������s�|���x����Ȼʹ�õ���ࡣͨ�^���Nɫ�V�����g�Լ��ü��g�܉^�����������F���x�����������˽�Q�c����z�y�����`���ȵ͵Ć��}�����Ͷ��S �� Ч Һ �� ɫ �V (2D-HPLC)������Һ��ɫ�V���g���x���������c����ˌ��͝���s�|�ęz�y������
Ŀǰ��HPLC �oՓ��������ϴÓ߀�Ƿ���ϴÓ�������Ĵ������ЙC�܄����@���h�������^�����Ⱦ����������ˮ�������h���Ѻõ�ԇ������ҪϴÓ�܄�������Һ��ɫ�V���g���õ��V�����о�������ЧҺ��ɫ�V���g(UHPLC) ���ˑ���ˎ���аl����Ҫ��UHPLC ����һ�N���ٷ��x��ɫ�V���g�ѽ����F�ڸ��Nˎ����_�l�ϡ�
Dong �ȸ���UHPLC ��ʹ�÷�����ӑՓ�����ᘌ���ͬ��Ʒ����������UHPLC���������Է��x���棬UHPLCĿǰ���]�д���С��2��m ��������Ʒ������ɫ�V�����������Â��y���������������ӷ�Ҳ�Ɍ��F�������s�|�M�з��x��
�ڌ���ҽ��O���棬���������Ŀǰ���� HPLC ����Ҫ�о��x���������˫@�������UHPLC ���xЧ�ܣ��S����й�����ͨ�^���� HPLCϵ�yͬ�rʹ�ú˚���ɫ�V�����F�˿��ٷ��x��
��2�����R�����wɫ�V(SFC)
��Һ�BCO2 ����Ҫ�������SFC ���g���������c�����Ùz�y�`���ȵͣ���lչһֱ�dz����������|�V�z�y��(MS)���ռ��Լ��h���Ѻ����������Ҫ��ʹ SFC �����ڷ��x�����s�|���mȻ SFC �c�|�V�ü��g�����s�|�����������P��� ������Ʒ���� SFC-MS�Ãx����δ�������F��
3���s�|�Ƃ��c�������g
�錍�F���s�|�Ķ��������Է�������Ҫ�@ȡ���Ȇ�һ�ɷֵ��s�|��Ȼ���s�|��ˎ���к����^�ͣ����÷�����Һ��ɫ�V���g�Ƃ��s�|��Ҫ���Ĵ����r�g�������Ƃ���Һ��ɫ�V���g�ȷ�������߸����s�|�ī@ȡ�ٶȣ��ӿ��s�|�о�������
��1���Ƃ���Һ��ɫ�V���g���Ƃ���SFC���g
���������s�|�ɫ@�ø����|���ĈD�V����ˎ�����s�|�����͵Ć��}һֱ�Ƽs���s�|���w�ī@ȡ�ٶȡ�����Һ�|�÷����ɫ@���s�|�ā�Դ�c���νY���������Ï��ƽ��� ���Y��ĸҺ��ֱ�Ӻϳɵȷ�ʽ���Ƃ���s�|���w�Ƃ似�g�ϡ�
���˿˷���Ҏһ�S�Ƃ���Һ��ɫ�V���g�Լ��Ƃ���SFC ɫ�V���g�ķ����������y���Ƃ�r�g�^�L��ȱ�c������Ƃ��͵Ķ��SҺ��ɫ�V���g(Prep 2D-LC)�Լ����S SFC ɫ�V���g�ѵõ�Խ��Խ��đ��á�
Zhang �������һ�N���͵� Prep 2D-LC��ԓ�x������ͨ�^һ�SҺ��ɫ�V����Ʒ�M�г������x�������|�V���������|���O�غ������и�ķ�ʽ��Ŀ���ﱣ���ژ�Ʒ�h(Sample loop)�У�����������ϴÓ(at column dilution)�ķ�������Ʒ�h�еĘ�Ʒ�M�����ڶ��SҺ��ɫ�V�У��ڶ��SҺ��ɫ�Vͨ�^ʹ���c��һ�S��ͬ���߲�ͬ������������Ʒ�M���Mһ�����xͬ�rʹ���|�V�O�ط������ɵõ������ȵ�Ŀ���
�@�N�·������H�����һ�S�Ƃ���Һ��ɫ�V��ͬ�r��Q�˂��y�Ƃ�2D-LC ϵ�y�߉��Լ���չ���Ć��}�������2D-LC��ԓ�n�}�MҲ�����2D-SFC �Ƃ似�g���������Ժͷ����Ի�����ĺY�x ��
��2�������Ƃ似�g
ˎ�����s�|�����ͣ�����Һ���������x�ķ�ʽ������s�|�pʧ�����L�Ƃ�r�g������ɫ�V���g ( counter-current chromatogra-phy��CCC)ͨ�^ҺҺ��ȡ��ʽʹ�s�|�����_����ͣ���Ʒ�جF���܉��_�� 100%�������Ј�����Ƃ�Һ��ɫ�V���g�� CCC �M�Ќ��ȣ��Y������ CCC���ܽ�ȵ͵Ę�Ʒ���Ϙ�������ͨ�������������Ƃ�Һ��ɫ�V��
�c CCC ��Ƶ��x�ķ���ɫ(centrifugal partition chromatography��CPC)���������w�o�����䷽ʽ���F��ˎ��ļ������ã���CPC ��ʹ���Е����F�̶�����ʧ�Լ����ٲ������}�_�Ć��}��Amarouche �� ͨ�^�������ϴÓ(co-current elution)������Q���������}���ɹ��،������ԭh���� A �M���˼�����
���ˌ��F�����Ƃ��c������ͨ�^�՚�Ӊ�����Һ�̷��x�Ŀ����|�V(flash chromatography��FC)Ҳ��ʧ��һ�N�����Ƃ仯����ķ�����
4���s�|�z�y���g
�mȻ HPLC-UV ���g�Ɍ���ˎ���s�|�M�ж�������������������z�y�`���ȵͶ��o�����F���O���s�|�Ĝʴ_������Ȼ�����|�V���g���и��`���Ⱥ߷ֱ��ʵȃ��c����������������Խ�Ķ����Ͷ��Է��������ѵõ��˿��ٵİlչ��ͬ�r���p�٘�Ʒ������Ҳ�Dz����ƄӺ˴Ź����g�İlչ����֮һ��
��1���|�V���g(MS)
�����������|�V���g����������o푑��s�|��һ�N��������ֶΣ�ͬ�r��������^�ߵęz�y�`���ȣ��܉�����β��L�z�y�o�������ĺ����s�|���Ԝʴ_�y�������ڌ�ijЩ�����s�|�y���r�������x�ӻ�����������Ҫͨ�^�������������������A�����x�ӵȷ����ԫ@ȡ�|�V푑� ��
��Ҏ MS�z�y�����ڷֱ��ʵ��Ƽs���������s�|�Ķ����ʴ_�Բ��ߣ����߷ֱ����|�V(HRMS)�܌���������ĺ��|��(m/z)�z�y�����`���1��10-6~2��10-6�����x���x�Ӓ��趨��������ˌ�����������Ķ����ʴ_�ȡ�
HRMS�߷ֱ��ʵ�����һ�����c���|���^�ָ���ʴ_���@�����څ^�����������|������Ķ�N��������ô˃��ݺ� UHPLC�����ڶ̕r�g�Ȍ��F����N�s�|�ķ��x���� ��
�Y���b�������s�|���w�M�нY���b���ĕr�L�������ˎ����s�|�V������ʹ��Һ�|�õķ����Ɍ��s�|�Y���M�п����b����ԓ��������һ���|�V�_���ķ����x�ӷ��M�ж����|�V���ѷ�����
Ȼ�����|�V�ֱ��ʵIJ��㳣ʹĸ�x�ӵ� m/z �Д�ʣ��Ķ�����s�|��Ԫ�ؽM�ɲ����_��ͬ�r������Ƭ��Ϣ������Ҳ��K�ˌ��s�|�Y�����Mһ����������ˣ�HRMS���༉�|�V(MS n )�Լ���/�(H/D)���Q�ȼ��g������Եă��c�܉��s�|�Y�������ʴ_�Ľ�����
�|�V�ֱ��ʵ�������������������|����Ϣ�Ĝʴ_�Բ��ɜʴ_�A�yԪ�ؽM�ɣ�ͬ�r�����|�V����ܛ��������Ӌ��ܛ�� ��Ӌ�����ͬ����ʽ�÷ָߵ͡�
�|�V�ֱ������Ҳ�Ѕ^�ֲ�ͬͬλ�صĹ��ܣ��������������|���� 500 ���ҵķ��ӣ�ֻ�зֱ����_�� 400000 ���܌��|�V�D�е�34S��37Cl�ɷNͬλ�ط���x�_��
��2���˴Ź����g(NM��)
NMR ���g���s�|���ԺͶ�����������Ҫ��ه�ګ@���s�|���w�����⣬�������s�|�|���˜ʽ����r���s�|����Ʒ�Ę˻������ö���NMR�M�� ��NMR���gͬ�rҲ��һ�N�|�����P�z�y���g��ʹ��NMR���g����У�������M��У�������z�y�����Ɍ��F�������M�̵ıO�ء�NMR���g�z�y�`������ه��̽ᘵ����ܡ�
��ˣ��������NMR�z�y�`���ȣ����о��߰l��������̽ᘣ��@�N̽�ֻ���˼��e�Ļ�������w���܌��F��������z�y���@ʹNMR�z�y��Ʒ���������F���ɺ������˵��w�S��ͬ�r�ھ���Һ��-�˴�(LC-NM��)�ü��gҲ�܌��Fˎ���s�|�Ŀ��ٽY���b�� ��
5�����͵ķ������g
�S�������s�|�����Լ��Y���b���ʴ_�Ե���Ҫ��Ҳ��һЩ�����������s�|�ĽY���b������ˎ���s�|ֱ�Ӝy�����g ���Է���ӡ�E��(MIP)����ͬ��s�|��Ƭ�������Լ��ξ� X �����似�g�ȡ�
�����������s�|�ķ���
�������s�|��ֱ�����������w�е� DNA����� DNA �p���������°����»�����ͻ׃�����|�������Ļ������s�|Ҳ�܌����w��ɘO��ēp�������������s�|���о���������ˎ���о��ߵďV���Pע ��
1����������
���ڻ������s�|Σ�U�ԘO������ FDA �͚W��ˎƷ������(EMA)Ҏ���˶���W���n�ֵ(TTC):�����L����ˎ�r�����ڶ����s�|ÿ�Քz�������ܳ��^1.5 ��g ��
��������Ҏ����ÿ��ˎ����Ä����� 200mg���t�����s�|���|���֔��������^7.5-��10-6 ����ˌ��������s�|����������Ҫ���������ķ�����
���õ�����z�y������܉��|���֔�5��10-4���s�|�M�Йz�y�����S�� 2D-HPLC�İl���Լ����`��������z�y����ʹ�ã�ˎ���к����s�|��һ�SҺ���x���x��������ȡ���g���������SҺ���x���`���șz�yҲ�܌��F�ʴ_������
���ǣ��@�N������Ҫ��θߝ���M�Ӳ����m���s�|�����y����
��ˣ��|�V�z�y���g�ǽ����������s�|����������������Ҫǰ�ᡣKakasaheb�� ������ɳ̹�кϳ���ʼԭ���еĻ������s�|���� GC-MS �ķ����M�ж����y���������W��C�Y���܉���� ICH Ҏ����Ҫ��ԓ�����m��������ˎƷ�л������s�|�ĺ����y����
�ں������s�|�M�з��������^���У���Ʒ��ǰ̎����ʽʮ����Ҫ�����ēpʧ���܌��Y����ɘO���Ӱ푣�Devenport �Ȅ����Ե��ڴ�≺�l���������|�Vֱ�Ӷ�����ģ�Mˎ���л������s�|���@�N����ʹ��Ʒ�y�����ӱ�ݣ����Ɍ��F��ˎ��ĸ�ͨ���z�y��
2�������s�|�Ĵ_
2006 �꣬EMA �l�����z�������ȵ�ָ��ԭ�t���˺� ICH��FDA �Լ��҇���ˎƷ���u����Ҳ����ˌ��������s�|���о��Ϳ��Ʒ������������P��ָ��ԭ�t����ʹ�s�|�ĺ�����Ҏ���Ķ���Ҫ������Ҳ��Ҫ���s�|�M�ж����u����
�������ﶾ���u����ͨ�^�w�⼚�����Ԍ��F�ģ�ͬ�rָ��ԭ�t���]ʹ���s�|���w�M���о���Ȼ����ˎ���еĶ����s�|��������������ڵģ�ͨ�^�Ƃ�ȷ����@ȡ�s�|���w�ĕr�L�ҳɱ��ߡ�
��ˣ�FDA ��������˾��ͬ�_�l��Ӌ��Cܛ�������u��������Ķ��ԡ���ܛ���o��@ȡ���w�����s�|�ĽY���M���b������ʹ�ö����u��ܛ���u���s�|�ĽY���Ƿ���ھ�ʾ�Y����
��ܛ�����棬FDA ���]ʹ��MC4PC��MDL-QSA�� �Լ� Derk for Window ���M���u�������ˌ����Ե��Ԝʴ_�Ե��u����ÿ�Nܛ������������㷨���m�÷�����
MC4PCܛ���nj����u���ĽY����ֳ� 2~3 ��(�ǚ�)ԭ�ӵĽY����Ƭ���ٌ��@Щ��Ƭ�c��֪���Ի�������Ƭ�������M�бȌ������s�|�Y���е���Ƭ�����˶����ۼӵ÷ֲ����L�Y���ķ�����������K����һ��ȫ����Ҍ��I�Ĉ���u���s�|�ĽY�����ԡ�
�mȻ MC4PC�܉���s�|�ľ����Y���Ƕ��u��ˎ��Ķ��ԣ�������ֻ�������еĔ�������������ͬ����Ƭ�M�б��^��������һ�Nδ֪��Ƭ�ĽY���s����ȫ���u����
���Ի�������о��Y���������Ի�����Y���ж��������H��ӻ��F��ɱ�������H��ӻ��F�ĽY����MDL-QSA�Ҷ����u��ܛ���ǻ��ڻ�����Ķ��Ժ�ԓ��������H����������P�������u�����@�Nܛ����Ӌ���������Y���c�䶾�ԵĶ����Pϵ��ͬ�r�����A�y�w�⼚�����Ԍ��ĽY����
Ȼ���������_������֪�Y�����s�|���Ե���ʴ_�u����ʹ�� MDL-QSA��ܛ���r�茤�ҵ����m�ϵ�ģ������Ӌ�㣬�����ԓӋ��ܛ����������S���P�ڲ�ͬ�Ĕ���ģ���Լ����������u���Ĉ����
��Щ�s�|�m��Ӌ��C�u���Y�����о�ʾ�Y���ĝ��ڶ����s�|���������u��ܛ����Ӌ���^���Е��^���u���s�|�Ķ��ԣ��������ж�ˎ���s�|�Ƿ�鶾���s�|����Ҫ�M���w�⼚���؏�ͻ׃���(Ames ���)������:��������FDA��ԓ�����^�̽o��Ԕ�����f����
3�����Ʒ���
���� EMA ָ��ԭ�t�Լ���ָ��ԭ�t���}���ˎ�������д��ڵĻ��������ԓͨ�^�����ﶾ���u�������ָ��ԭ�t�Ŀ��Ʒ�����ͬ�r���˴_�����ڶ����s�|�Ƿ�鶾���s�|��Ҫ�M��Ames��
�ġ�չ��
�s�|�о���ˎ���аl�^����ռ�м�����Ҫ�ĵ�λ�����HӰ�����ˎ���е��ٶȣ���Ӱ�����ˎ��ȫ���S���������g�IJ���lչ���s�|�о��IJ����ѽ�׃�����l���ӻ����S���z�y���ֱ����Լ��`���ȵ���ߣ��s�|�������F���������������w�S���s�|�Ķ��Թ���Ҳ���F�����x�����ھ��z�y���D׃��Ȼ������ˎ�аl�ٶȵļӿ죬����ˎ��IJ���ӿ�F�Լ����ٶ��Զ���������Ҫ���o�о��ˆT����O�������